
Patient Information
Cystinosis is also referred to as Nephropathic Cystinosis. This emphasises its effects on the kidneys and distinguishes it from another form of Cystinosis that only affects the eyes. Note that Cystinosis must not be confused with Cystinuria which is an entirely different condition.
Information about Cystinosis national designation centres or “hubs” for patients in England
The first signs of Cystinosis usually begin between 3-18 months of age. Boys and girls are affected equally. One of the first signs of Cystinosis is that the child becomes more and more difficult to feed. They are thirsty but have poor appetite. Their growth slows and they develop muscular weakness.
Affected children are very vulnerable to dehydration. For example a minor episode of diarrhoea or vomiting that would pose very little risk to healthy child may cause severe dehydration. Also, in a child with Cystinosis the clinical signs of dehydration are more difficult to recognise. This means that by the time a parent or a clinician recognises the problem it may be serious. (To access a Medical Alert click here)
Young children with Cystinosis also develop softening of bones (rickets), even if their diet contains normal amounts of Vitamin D. Bones can become painful so that walking may be delayed and a small child stops using a limb or cries on being picked up.
Cystinosis also affects the eyes. Children with Cystinosis hate bright light, and squint or turn away from it (photophobia). The accuracy of their vision is not affected. They often need to wear dark glasses when they’re outdoors.
The disease is progressive and without corrective treatment children lose kidney function in about 10 years. Treatment can extend kidney function beyond childhood, and physical strength and growth are improved. However, there is no permanent cure for the condition. Later on patients will require renal replacement treatment – dialysis or a kidney transplant. Cystinosis does not come back in the transplanted organ so transplantation is usually successful.
Complications can develop in adulthood due to the long term build-up of cystine in the organs. Potential problems include diabetes, muscle weakness, thyroid problems and weak bones.
The Cystinosis Rare Disease Group has written an Information Prescription for patients with Cystinosis. For further details and to download the document please click here.
The first step is to confirm the diagnosis. In Cystinosis there is a build up of cystine in the body. Cystine is a naturally occurring amino acid (see How the disease works below). The diagnosis is made using a blood test that measures the amount of cystine in white blood cells. For more information about the blood tests click here.
Different treatments are needed at different stages of the condition.
In very young children a major issue is keeping them well hydrated and nourished.
Because in Cystinosis the kidneys leak essential salt, water, potassium and phosphorus, all of these have to be given as a nutritional supplement. Kidneys also regulate the balance between acid and alkali in the body. Also excessive amounts of bicarbonate (alkali) are lost in the urine leaving too much acid behind in the patient. The excess acid makes appetite and growth worse. The control of these aspects is crucial and requires frequent hospital visits and blood tests. Medical care should be obtained from a specialised paediatric nephrology service. For a full list of UK paediatric nephrology centres who have experience of this disease click here.
As children get older the risk of dehydration gets less. The need for replacement potassium and phosphorus reduces or stops. However, nutrition and growth require ongoing supervision. On average children with Cystinosis grow less well than normal children and go into puberty later. Most require a supplement of thyroid hormones, thyroxine, as their thyroid gland is under active. Some may benefit from growth hormone to increase growth rate before puberty.
Specific treatment
At all stages of the disease the build up of excess cystine in the body can be controlled using Cysteamine. Cysteamine is unpleasant to take by mouth due to its sulphury smell like rotten eggs. Cystagon™ is cysteamine bitartrate, a pro-drug which is converted to active cysteamine in the body after being swallowed. It is better tolerated than cysteamine which is why it’s preferred.
Cysteamine eye drops can also be used to clear the front part of the eye, the conjunctiva and cornea.
These treatments have radically improved the outcome of Cystinosis and may delay damage to other organs. Good medical supervision and strict adherence to treatment significantly delays the time when dialysis or a kidney transplant are needed.
Patients who develop additional problems such as diabetes or muscle weakness will need to be treated by other specialists. This could include a neurologist (nervous system) and an endocrinologist (hormones).
The Cystinosis Life website features four young patients telling their stories in their own words. Topics covered include friends and family, everyday life and transition to adult services.
The Cystinosis Foundation UK supports individuals, families and researchers in the Cystinosis community.
Cystine is an amino acid, a building block of protein. In the body many different proteins need cystine to give them their specific structure. Cystine is commonly found in the skeleton, connective tissue, hair, skin and nails. The body can make its own supply of cystine from other amino acids. Two cysteine molecules joined together make one cystine molecule.
Proteins are broken down in every cell of the body and the amino acids are mostly recycled. This takes place in a lysosome, a microscopic bag-like structure inside the cell.
In Cystinosis, cystine released inside the lysosome cannot get out into the other parts of the cell. This is because the special transport system in the wall of the lysosome is faulty. As a result cystine builds up in the lysosome. Cystinosis is one of a group of metabolic diseases known as lysosomal storage disorders. Cystine doesn’t dissolve very well in water and forms crystals in the body. This can be seen with a microscope in certain tissues.
Cysteamine, the basic treatment of this condition, dissolves very well in water and gets into the lysosome of the cell. Here it reacts with cystine, splitting the molecule. This produces cysteine (effectively half a cystine molecule) and another soluble product (cysteamine-cysteine disulphide). These do not need the same transport system so the cystine is effectively removed.
The two parts of the body most affected by cystine overload are the kidneys and the eyes. In the kidney the build up of cystine occurs mostly in the cells of the proximal tubule. This is the part of the kidney responsible for reabsorbing large amounts of salt, water and phosphorus. This is why in its early stages Cystinosis is characterised by an excessive loss of fluid and the presence of salt and phosphorus in the urine. This pattern of tubular malfunction is also known as the Renal Fanconi Syndrome. Later on kidneys become extensively damaged and scarred so that overall kidney function is lost.
In the eye, cystine crystals are deposited in several areas including the cornea which is the cause of photophobia. Here the crystals can be seen by examining the eye with a slit lamp.
How is the disease inherited? Cystinosis is a genetic disorder and is inherited.
The gene involved in Cystinosis is called CTNS and it affects a protein called cystinosin. Cystinosin is a transport protein and carries cystine out of the lysosome. We have two copies of this gene, one inherited from each parent. In order for a patient to develop Cystinosis both copies of the gene must be faulty.
Parents of a person with Cystinosis are almost certain to carry one copy of the faulty gene. Because they have a normal copy as well, they are themselves unaffected. One normal copy of CTNS is sufficient to produce enough normal cystinosin for normal cellular function. Parents are therefore carriers of the condition. This pattern of inheritance from unaffected carrier parents to affected child is known as autosomal recessive inheritance.
This diagram shows how Cystinosis can be passed on to the children of two carrier parents.
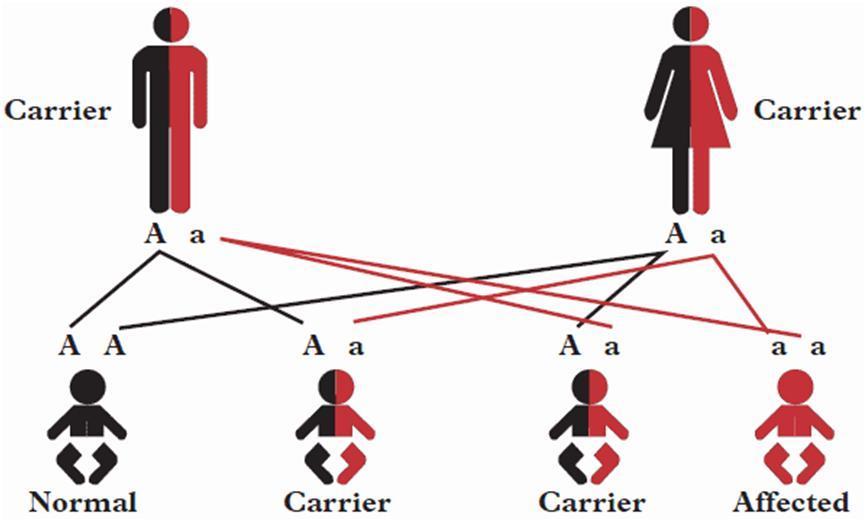
A = unaffected gene a = affected gene
The Cystinosis Rare Disease Group (RDG) is working with international partners with the aim of finding new and improved treatments, and to empower patients. A first step is to compare the symptoms and genetic markers of Cystinosis. To do this the RDG is registering patients with this condition in the National Renal Rare Disease Registry (RaDaR).The database will be used to find suitable participants for future research trials into the effectiveness of new treatments. If you are interested in finding out more about the registry RaDaR or the activity of the RDG please visit the Cystinosis RDG page. Delayed release Cysteamine (Procysbi™) is a form of cysteamine bitartrate that needs to be taken every 12 hours. It is as effective as Cystagon™ taken every 6 hours. It has been launched in the USA and has been licensed by the European Medicines Association but is not yet available on the NHS.
- The Cystinosis Foundation UK supports individuals, families and researchers in the UK Cystinosis community.
- The Cystinosis Foundation is the US equivalent.
- The Cystinosis Research Network is dedicated to supporting research and educating the public and medical communities about Cystinosis.
- Rare Disease UK is the national alliance for people with rare diseases and those who support them.
- Climb (Children Living with Inherited Metabolic Diseases) provides Metabolic Disease specific information, advice and support to children, young people, families and professionals in the UK.


